720-777-0123
720-777-0123







Key takeaways
-
Treatment with LUM/IVA improved growth and reduced sweat chloride levels in a F508del homozygous population. The study offers the most robust estimate to date in this population of LUM/IVA’s effects on sweat chloride changes, an important measure of CFTR function.
-
Younger participants had the most gains in growth, suggesting that starting treatment with LUM/IVA at a young age may yield the most benefits.
-
In contrast with the phase three clinical trials, treatment with LUM/IVA did not significantly improve lung function in this real-world cohort.
Background: effect of lumacaftor/ivacaftor on clinical outcomes in a cohort of patients homozygous for the F508del mutation
A key approach to treating cystic fibrosis (CF) is the use of small molecule therapies designed to improve the activity of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, the protein in the body that does not function normally in people with CF.
The most common mutation in the gene that produces the CFTR protein is F508del. Almost 50% of people with CF in the U.S. are homozygous for this mutation (have two copies inherited from both parents). In 2015, the U.S. Food and Drug Administration (FDA) approved a medication for this population that combines two drugs, lumacaftor (LUM) and ivacaftor (IVA). LUM helps the CFTR protein fold into a more stable shape so that more of this protein is transported to the cell surface and IVA allows more chloride to move through the CFTR channel on cell surfaces.
The large multinational clinical trials that led to this combination medication’s approval showed that, in people with CF homozygous for F508del, this treatment modestly improved lung function, growth and quality of life. It also significantly reduced pulmonary exacerbations (respiratory illnesses treated with antibiotic therapy).
European studies of real-world, post-approval experience with LUM/IVA have found many patients discontinued the drug due to adverse side effects. Those investigations found people who continued treatment benefited similarly to those in the registrational trials.
Scott D. Sagel, MD, PhD, and other Children’s Hospital Colorado researchers, led a multicenter study to examine the real-world clinical effectiveness of LUM/IVA in the U.S. population. Dr. Sagel is a pediatric pulmonologist in the Mike McMorris Cystic Fibrosis Research and Care Center within the Breathing Institute at Children’s Colorado and director of the University of Colorado Cystic Fibrosis Center.
The researchers reported on outcomes in children and adults homozygous for the F508del-CFTR mutation before and up to one year after starting the medication. Another purpose of this study was to perform investigations in a subset of study participants to examine the effects of LUM/IVA on lung ventilation, mucus clearance from the lungs, digestive health and insulin secretion.
Methods: multicenter longitudinal cohort study evaluating clinical outcomes with LUM/IVA
Researchers at 38 centers in the U.S. CF Therapeutics Development Network enrolled patients aged 6 years and older with two F508del CFTR mutations and no prior exposure to LUM/IVA.
Participants were assessed at baseline and 1, 3, 6 and 12 months after they began LUM/IVA therapy for:
- Lung function (percent predicted forced expiratory volume in 1 second [ppFEV1])
- Sweat chloride, an indicator of CFTR protein activity
- Quality of life survey
- Height
- Weight
At baseline and one year after LUM/IVA initiation, researchers reviewed patient data on:
- Hospitalizations for pulmonary exacerbations
- Pseudomonas aeruginosa infection based on respiratory cultures
Samples of participants’ blood, urine and induced sputum, and at some sites, a one-time collection of participants’ nasal and epithelial cells were collected for future study.
Results: improved growth and reduced sweat chloride levels with no significant changes in lung function, hospitalizations or infections
Of the 193 study participants who started therapy with LUM/IVA, 90% completed study visits through six months and 85% completed visits through one year.
Study participant baseline characteristics
The cohort’s baseline lung function was high (mean ppFEV1 = 85, SD=22.4).


Results of core clinical outcome measures
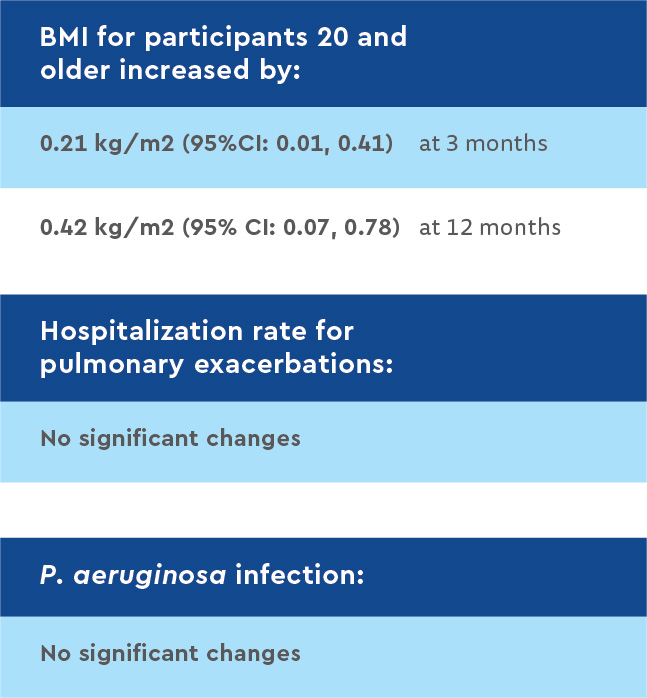
ppFEV1: No statistically significant change in ppFEV1 from baseline to any follow-up time points
Sweat chloride levels: Reduced -18.5 mmol/L (95%CI: - 20.7, -16.3) from a baseline mean of 100.4 (SD=11.5) to a one-month mean of 81.6 mmol/L; reductions lasted throughout the study period.


Clinical outcome measures by age and baseline ppFEV1 category

Discussion and conclusion: improved growth and sustainably reduced sweat chloride levels
This post-approval observational study showed treatment with LUM/IVA significantly improved growth and reduced sweat chloride levels. Treatment did not lead to significant changes in lung function or in rates of pulmonary exacerbations or P. aeruginosa infections.
Investigators found significant reductions in sweat chloride levels at one month. These reductions were sustained throughout the one-year study period, confirming the bioactivity of the combination drug and the improvement in CFTR protein activity. According to researchers, this study offers the most robust estimate to date in a F508del homozygous population of LUM/IVA’s effects on sweat chloride changes, an important marker of CFTR function.
In contrast to the large phase three clinical trials, this cohort experienced no significant improvement in lung function. The difference may be due to this cohort’s higher baseline lung function that limited the ability to detect further improvements compared with previously studied populations.
The lack of lung function improvement in this real-world cohort also highlights the heterogeneity in clinical response to LUM/IVA among F508del homozygous CF patients. Participants’ nutritional status, based on increases in body weight, improved steadily over the 12-month study. Since the beneficial effects of LUM/IVA on nutritional status were most evident in children – who have the most growth potential – CFTR modulators may be particularly beneficial if begun early in life.
These results justify further development of more effective CFTR modulator combinations, as recently accomplished with the triple combination (elexacaftor-tezacaftor-ivacaftor) therapy.
Ongoing analysis of airway and circulating biomarkers and final analysis of several associated sub-studies may reveal additional mechanistic insights into partial correction of the F508del protein.
Featured Researchers

Scott Sagel, MD, PhD
Pulmonologist
Children's Hospital Colorado
Professor
Pulmonary Medicine - Department of Pediatrics
University of Colorado